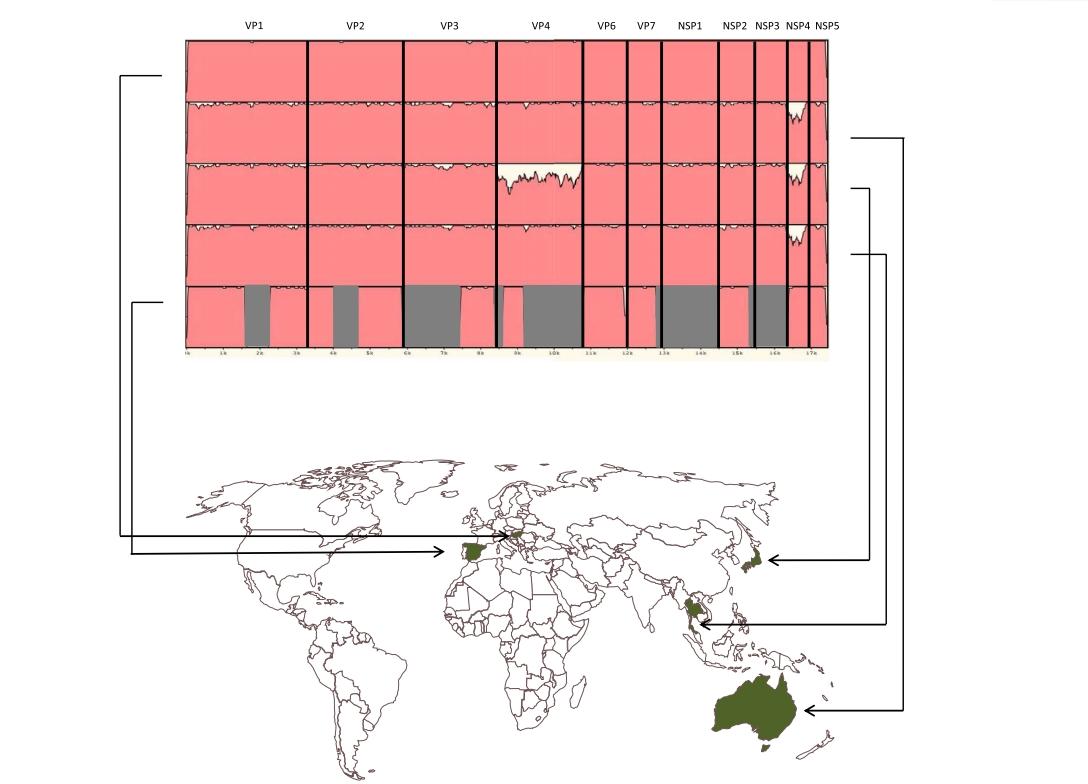
Whole genome based surveillance of rotaviruses: a new model for molecular epidemiological investigations
Rotavirus strain surveillance has been launched recently in numerous countries to support ongoing or anticipated vaccination programs. In Hungary, rotavirus surveillance dates back to the pre rotavirus vaccine era and has been conducted uninterruptedly over the last 20 years using standard typing methodology. The whole genome sequence based surveillance of rotaviruses would be undertaken in order to gather comprehensive information on the rotavirus genomic diversity in Hungary encompassing at least 9 consecutive rotavirus seasons, from 2006/2007 to 2014/2015 in selected surveillance areas of the Hungarian rotavirus strain monitoring network. The main objectives of the study are: (i) to describe in detail the molecular epidemiology of rotavirus infections in Hungary, during consecutive rotavirus seasons at a depth that could not be imagined until recently; (ii) to monitor the emergence and spread of common and novel rotavirus strains in Hungary; (iii) to monitor the possible vaccine-induced emergence of antibody escape mutants; (iv) to identify the possible emergence in the general population of reassortants between vaccine and naturally circulating wild-type strains; (v) to identify revertant vaccine strains that causes disease and spread from vaccinees to siblings or other susceptible individuals; (vi) to trace the evolution of rotaviruses in immunocompetent and immunocompromised patients.
There is a consensus that with the availability of rotavirus vaccines throughout the world, continuation of strain surveillance in the future will be required. This post-vaccine strain surveillance is facing several new challenges. To improve data quality surveillance should be standardized. Sufficient numbers of samples to be able to identify potential vaccine driven events (eg., vaccine breakthrough strains, reassortment events between vaccine and wild type strains) should be characterized. To help with this effort, typing methods need to be standardized across laboratories to minimize inter-laboratory differences. These changes will be critical to precisely assess the vaccine efficacy against various strains and document any changes in strain prevalence associated with increased vaccine use. The primary research questions and main challenges are: How could a whole genome sequence based surveillance substitute the standard techniques in this era? Is it feasible? Is it cost-effective? Does it provide the amount of information needed to adequately support existing vaccination programs? Is it suitable to recognize the main evolutionary mechanisms and explain the complex epidemiologic features of rotavirus?
The perspectives to investigate viral molecular epidemiology based on whole genome sequences has been recognized many years ago, however, very few studies have been carried out to date. Among vaccine preventable viral infections, the most intensive whole genome based characterization has been carried out with the influenza viruses. In other fields of virology, molecular epidemiological investigations mostly rely on the analysis of partial gene sequences; these fragments are often shorter than 1% of the whole genome, thus masking potentially relevant molecular changes in other genomic regions and permitting only limited conclusions on epidemiological features and evolutionary mechanisms to be made. The significance and novelty of the present project is that a whole genome based surveillance for rotavirus, a globally important vaccine preventable infection, has not been initiated anywhere in the world. Due to the ability of rotaviral genomes to undergo zoonotic transmission, reassort or recombine, accumulate mutations, duplicate or lose parts of the genome, whole genome based sequencing could be the best methodological approach during the vaccine era. This is the only approach that may uncover rotavirus evolution at the desired depth and could help understand the genes other than VP4 and VP7 that are important in immunity to rotavirus. Also, a better understanding of the evolutionary mechanisms is key to design more efficacious and safer vaccines.
Bayer-Dandár Eszter, biológus, Egyesített Szent István és Szent László Kórház-Rendelőintézet, Budapest
Dr. Antalné Dr. László Brigitta, PhD, Orvosi Mikrobiológia, Orvos- és Egészségtudományi Centrum, Debreceni Egyetem